
论文笔记(Learning functional properties of proteins with language models)
Published:
蛋白质数据的表征,是解决当前生物医学问题的一大关键,比较好的方法是从优秀的语言模型汲取灵感,以构建类似的生信模型,这些模型最近已经在序列-结构-功能关系提取上获得了很大的成功。这篇论文对蛋白质表征学习做了深入的调研,分类并解释每一种方法,依次在如下预测目标中对他们进行评估:
- 背景知识
- Overview
- 语义相似性推理
- 3.1. 概述
- 3.2. 过程
- 3.3. 结果
- 3.4. 关键点:
- Ontology-based蛋白质功能预测
- 基于药物的蛋白分类
- 蛋白质-蛋白质亲和性估计
Ontology(https://en.wikipedia.org/wiki/Ontology_(information_science)) 在计算机科学和信息科学中,本体是指对概念、数据和实体之间的类别、属性和关系的表示、命名和定义,这些概念、数据和实体构成了一个、大量或所有的论域[1]。本体提供的是特定领域之中那些存在着的对象类型或概念及其属性和相互关系[2];或者说,本体就是一种特殊类型的术语集,具有结构化的特点,且更加适合于在计算机系统之中使用;或者说,本体实际上就是“对特定领域之中某套概念及其相互之间关系的形式化表达(formal representation)” Ontology-based data integration(https://en.wikipedia.org/wiki/Ontology-based_data_integration) 使用一个或更多ontology去整合大量混杂的数据信息
1. 背景知识
截至2021年5月,UniProt收录了大约215,000,000条蛋白质序列,然而只有560,000(~0.26%)条序列被人工检查、标记过。这是因为标记需要大量cost。为了进行自动标记,很多团队开始研究新的计算方法以预测蛋白质的:
- 酶活动
- 生物物理学特性
- 蛋白质-受体交互
- 3D结构
- 各种功能
蛋白质功能预测(Protein function prediction, PFP)是蛋白质功能对蛋白质的自动化或半自动化的assignment。Gene Ontology(GO)系统是一个层次的概念网络,代表着从基因到蛋白质的映射,其底层逻辑则是蛋白质亚细胞定位和生物逻辑过程。
PFP任务中最具综合性的benchmark是Critical Assessment of Functional Annotation(CAFA),需要对一个目标蛋白质set预测他们基于GO的功能联系,然后再进行人工检验核算结果。
2. Overview
对不同方法根据技术特征和其application分类,然后进行评估。具体评估任务涵盖了:
- 背景知识
- Overview
- 语义相似性推理
- 3.1. 概述
- 3.2. 过程
- 3.3. 结果
- 3.4. 关键点:
- Ontology-based蛋白质功能预测
- 基于药物的蛋白分类
- 蛋白质-蛋白质亲和性估计
这篇论文选取了23种方法作为benchmarking task,后面的介绍将包括每种方法protein representation的构建和应用,以及对应的下游任务和评估。本文一共比较了39种方法(包括前面的benchmark),这些方法的平均表现总结如下表:
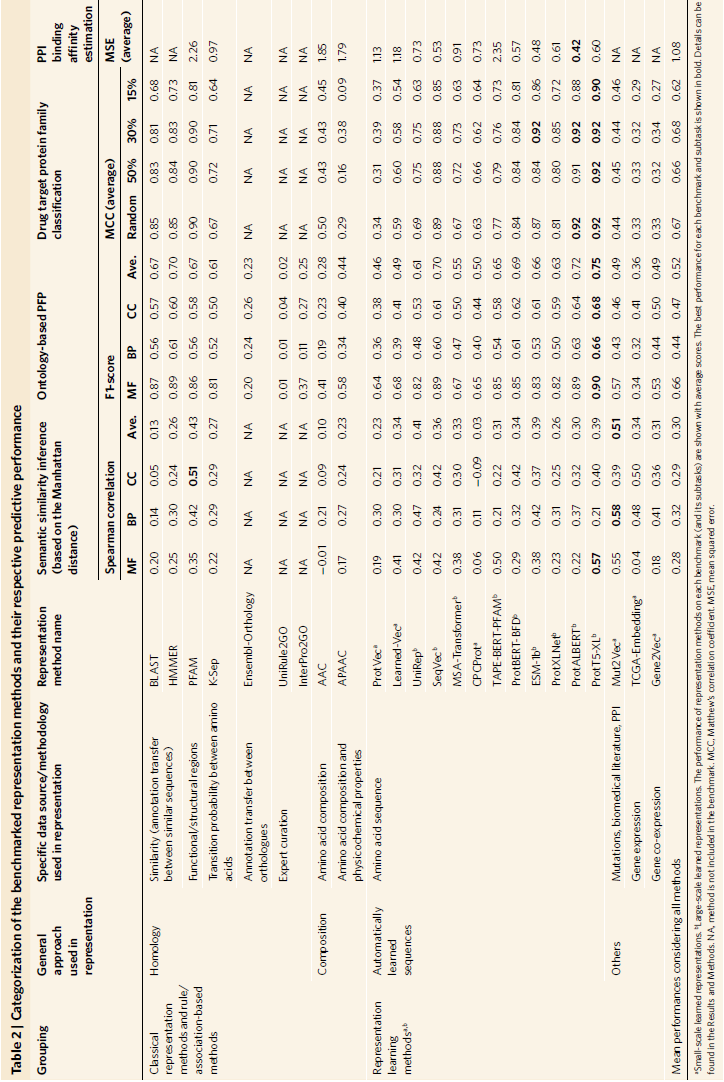
(有的method对于特定的任务并不适用,所以在表中会有一些NA)
蛋白质表征学习可以分为两个类别:
- 蛋白质级别的学习
- 氨基酸残基级别的学习
这篇论文主要讨论的主要是蛋白质级别的学习(除了最后一个任务,突变对蛋白质-蛋白质亲和性的改变)
3. 语义相似性推理
3.1. 概述
这个任务主要想衡量模型捕捉生物分子的功能相似性(biomolecular functional similarity)的能力。故而实验中采用了GO annotations以表征分子级别的功能、大规模的生物逻辑角色和亚细胞定位。
3.2. 过程
首先,使用曼哈顿距离和欧几里得距离定量计算representation vector的相似性。
曼哈顿距离(Manhattan distance/Manhattan length):标明两个点上在标准坐标系上的绝对轴距之总和。又称计程车几何(Taxicab geometry),方格线距离
计算公式:$d(x, y)=|x_1-x_2|+|y_1-y_2|$ 曼哈顿距离依赖坐标系统的旋转,不依赖坐标系统的平移
然后,通过真实的GO annotation和标准的比较语义相似性的方法(比如Lin similarity)比较这些蛋白质ground truth之间的相似性。
Lin similarity: 由于annotation更多有复杂的网络层级结构,所以直接比较相似程度没有从语义上出发来得准确。 一个单词可以表示一个类别。对于两个类别C和C’,它们的信息量(信息熵)之和可以表示为: -logP(C)-logP(C’) P(C)代表在整个样本空间(单词样本空间)中随机采样采到C类样本的概率 而这些类别之间,往往有着复杂的层级结构,他们可能有多个公共父类,越顶层的父类,往往越抽象;越底层的父类,往往越具体。这些父类中和这两类有着最近、最直接从属关系的,应当是我们最需要关注的。如果两个类在语义上越相似,那么最具体的那个父类所表达的信息就越能涵盖这两个类别的信息。所以可以定义如下相似性: sim(x_1, x_2)=\frac{2\times logP(C_0)}{logP(C_1)+logP(C_2)} 例如:在这个词汇网络中:
sim(hill, coast)=\frac{2\times logP(geological-formation)}{logP(hill)+logP(coast)}
最后,通过representation similarity和ground truth GO similarity的Spearman rank-order correlation value来评估representation的好坏。correlation value越大,表示representation越好。
在统计学中,斯皮尔曼等级相关系数(简称等级相关系数,或称秩相关系数,英语:Spearman’s rank correlation coefficient或Spearman’s $\rho$)常以希腊字母$\rho$或以$r_{s}$表示,这一相关系数以查尔斯·斯皮尔曼(英语:Charles Spearman)之名命名。它是衡量两个变量的相关性的非参数指标。它利用单调函数评价两个统计变量的相关性。若数据中没有重复值,且当两变量完全单调相关时,斯皮尔曼相关系数为+1或−1。 计算公式:$r_s=\rho_{R(X),R(Y)}=\frac{cov(R(x), R(Y))}{\sigma_{R(X)}\sigma_{R(Y)}}$ R表示将一组原始数据X转换成等级数据R(X)的函数
3.3. 结果
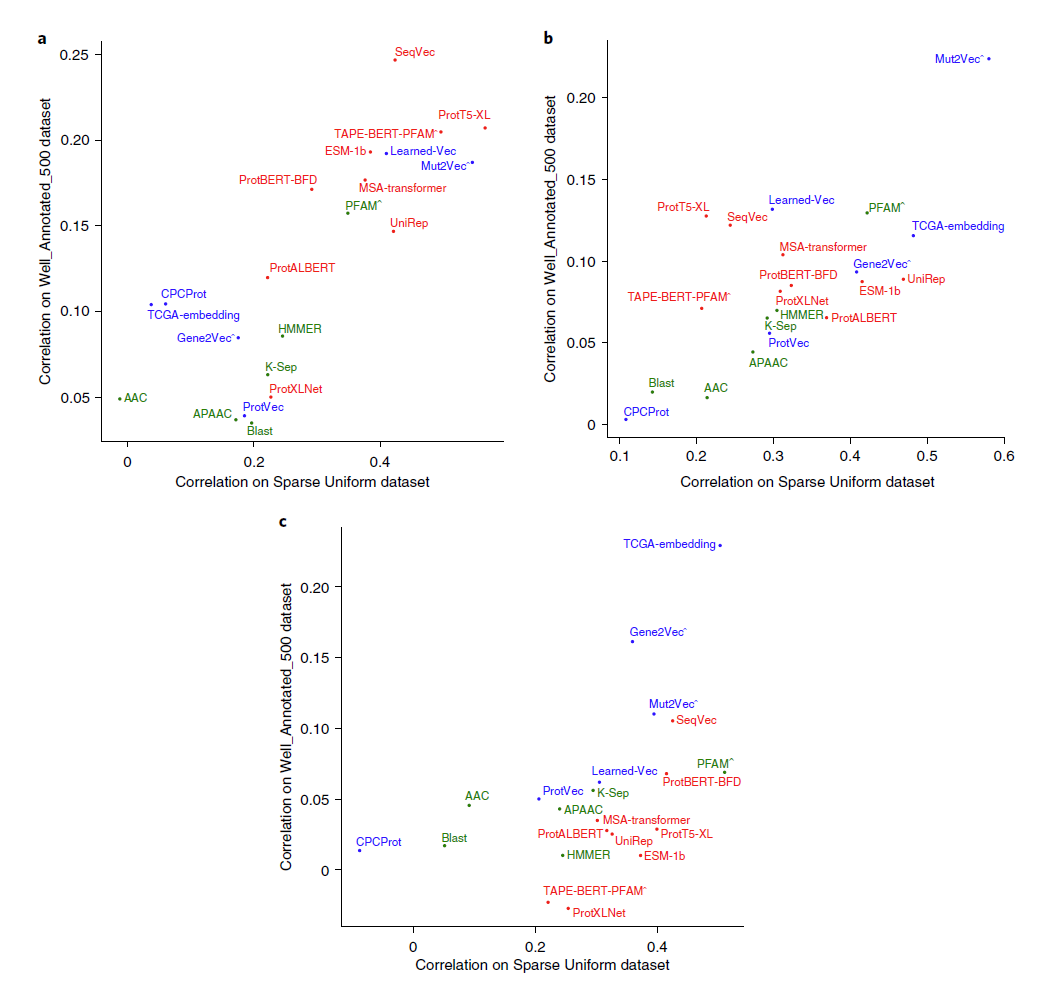
a,b,c代表不同的GO categories:
- a: molecular function, MF
- b: biological process, BP
- c: cellular component, CC
每张图的一个点代表一个模型的表现,横坐标值表示在Sparse Uniform数据集上的correlation,纵坐标值表示在Well Annotated 500数据集上的correlation,点的位置越靠右上越好。
具体计算方法:
- ground truth GO的similarity list: 用Lin’s measure
- representation的similarity list: 用曼哈顿距离
- 两个list的相关性:Spearman correlation
点的颜色表示模型规模:
- 绿色:经典representation
- 蓝色:小规模学习得到的representation
- 红色:大规模学习得到的representation
3.4. 关键点:
- 数据(人类蛋白质组)非常庞大而稀疏,不同的methods表现起来差别不大。
- distance metric对结果影响很大。
Ontology-based蛋白质功能预测
这一个task做的事情就是预测GO term annotation,本质上是一个分类任务。在representation后加一个linear分类器即可。
如果用更加复杂的分类器,那么就无法判断结果是因为representation足够好还是分类器足够好了。用简单的线性分类器则可以证明representation达到了线性可分的状态,是足够好的。
这篇论文中,作者关注了GO term之间的关系和每个term的信息量。比如,GO term “negative regulation of molecular function”这个annotation太general了,在GO graph中往往接近graph的root,并且含有较少的信息量,作者称这类term为shallow terms。如果是GO term “negative regulation of double-stranded telomeric DNA binding”这个annotation则会更具信息量。因此,作者把所有的GO term根据他们在GO graph中的深度分为shallow, normal和specific这三类。
此外,作者还根据带有这个annotation的protein的数量将annotation分类(low, middle, high)。这十分有意义:
- 揭示训练数据很少的时候结果如何。很多情况下,非常具体的annotation所对应的蛋白质会很少。
- 有的function被研究的很透彻,annotation很多,有的function还待研究(under-studied),作者expect语言模型对于under-studied的function能有很好的表征能力(若真如此,语言模型会对研究这些新功能非常有意义)
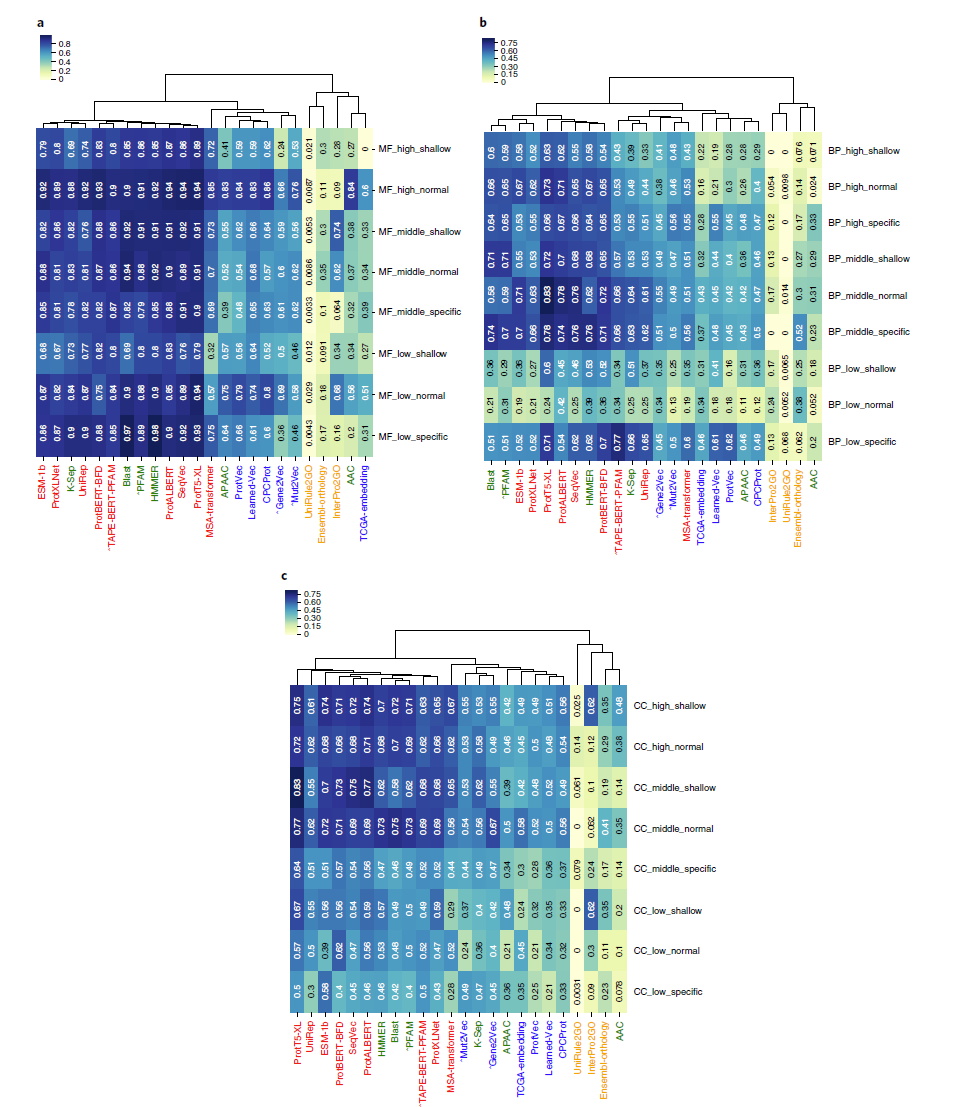
GO group一共有9类($[low, middle, high]\times[shallow, normal, specific]$),结果用F1-score-based热力图展示。
整体的结果(包括recall,precision,F1-score,accuracy, Hamming distance)如下表。由于建模方式特殊,这篇文章的结果比CAFA challenges中报告的结果要好。不过,作者主要的意图是从不同角度比较各个语言模型的表现,而不是选出最佳的一个来做PFP(后者是CAFA challenge的目标)。
MF 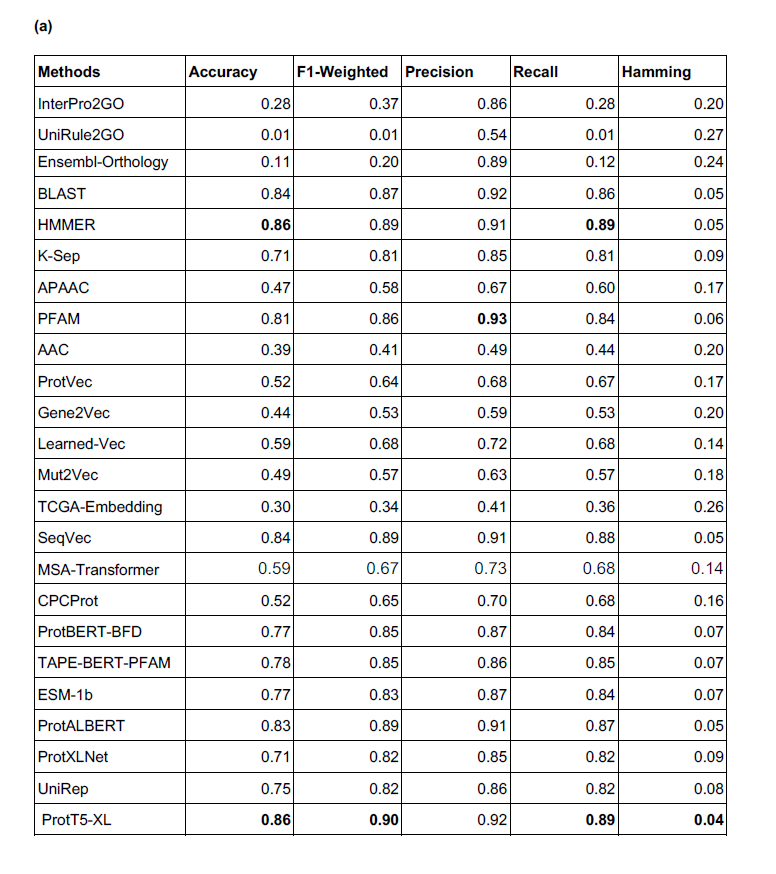
BP 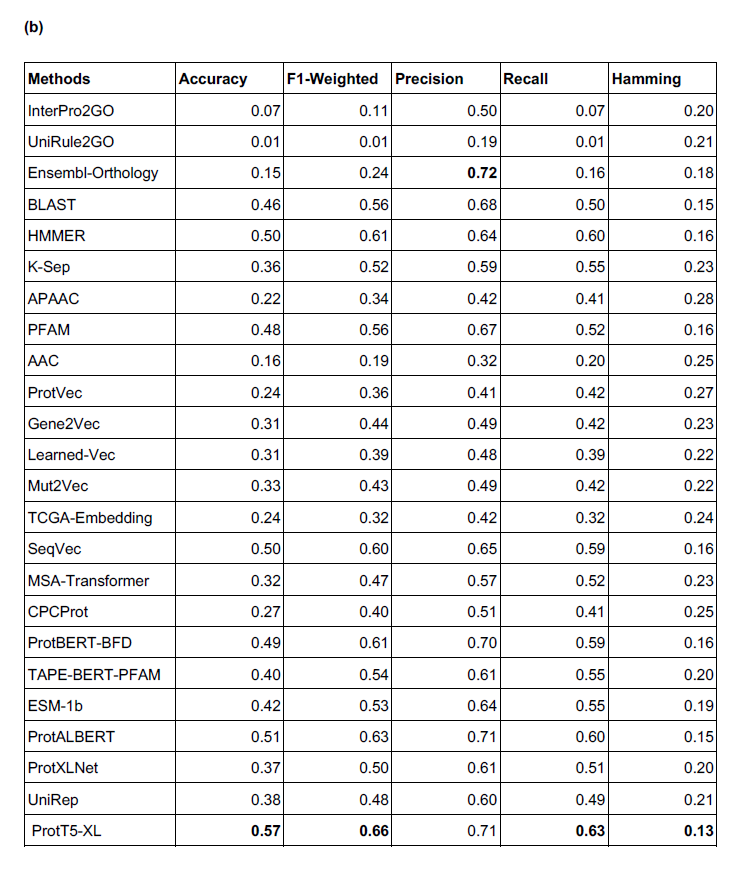
CC 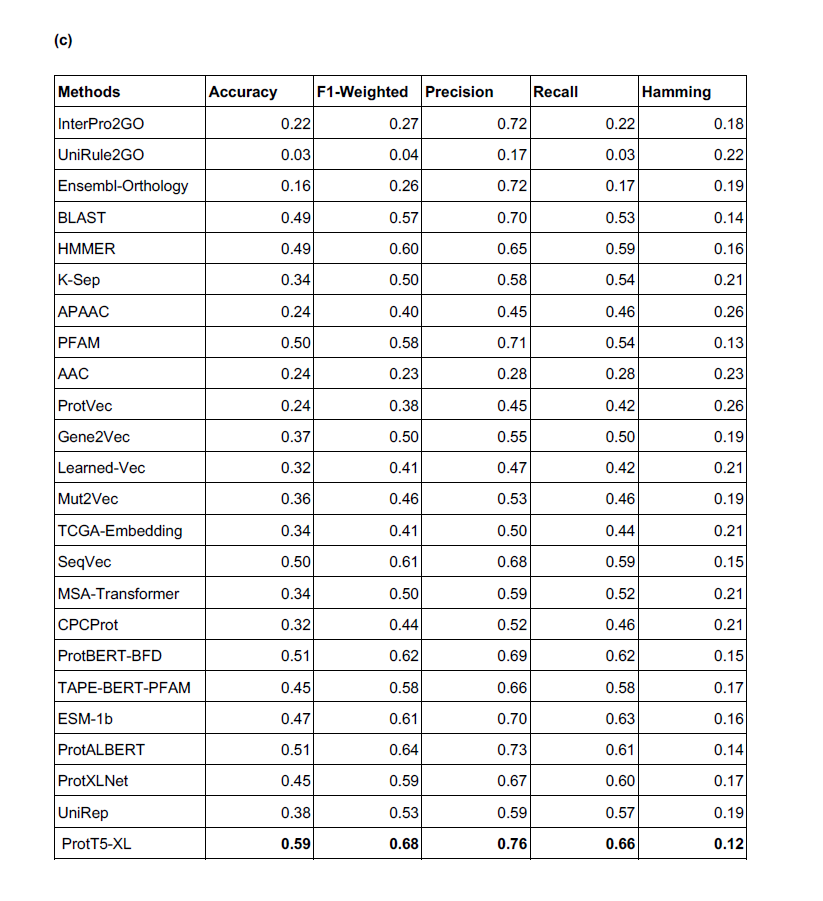
CC和BO两类annotation的预测结果比MF稍差,这可能是因为输入只有序列数据,而序列和localization(某个cleaved signal peptides可能在结构中不存在)并没有直接的联系,从而也和蛋白质在一个很大规模的生化过程中起到的生物逻辑学作用没有直接关系。
对于数据量和结果好坏的关系,CC term的结果随着数据量减小变差的比较明显,MF和BP则只有略微的变差。
对于term specificities(shallow/generic terms v.s. specific/informative term),并没有观察到显著的差异。然而,仍然可以认为specific/informative的GO term是有一定区别的,毕竟它们对应的数据量有显著差异。
基于药物的蛋白分类
从药物发现的角度看,蛋白质可以分为几个大家族:
- 酶 enzymes
- 膜蛋白/膜上的receptor membrane receptors
- 转录因子 transcription factors
- 金属离子通道 ion channels
- 其他
正是蛋白质的这层属性,决定了蛋白质结构上的功能。所以这项指标也能反映模型学习结构特性的能力,这样也能丰富评价指标,从另一个角度来评估representations。
完成这项任务的数据集共有4个版本:
- 随机划分的数据集
- 50%相似 (Uniclust50)
- 30%相似 (Uniclust30)
- 15%相似 (MMSEQ-15 clustering)
这样划分是为了研究similarity对学习蛋白族的影响。这些数据集做了tenfold cross-validation。
结果如下:
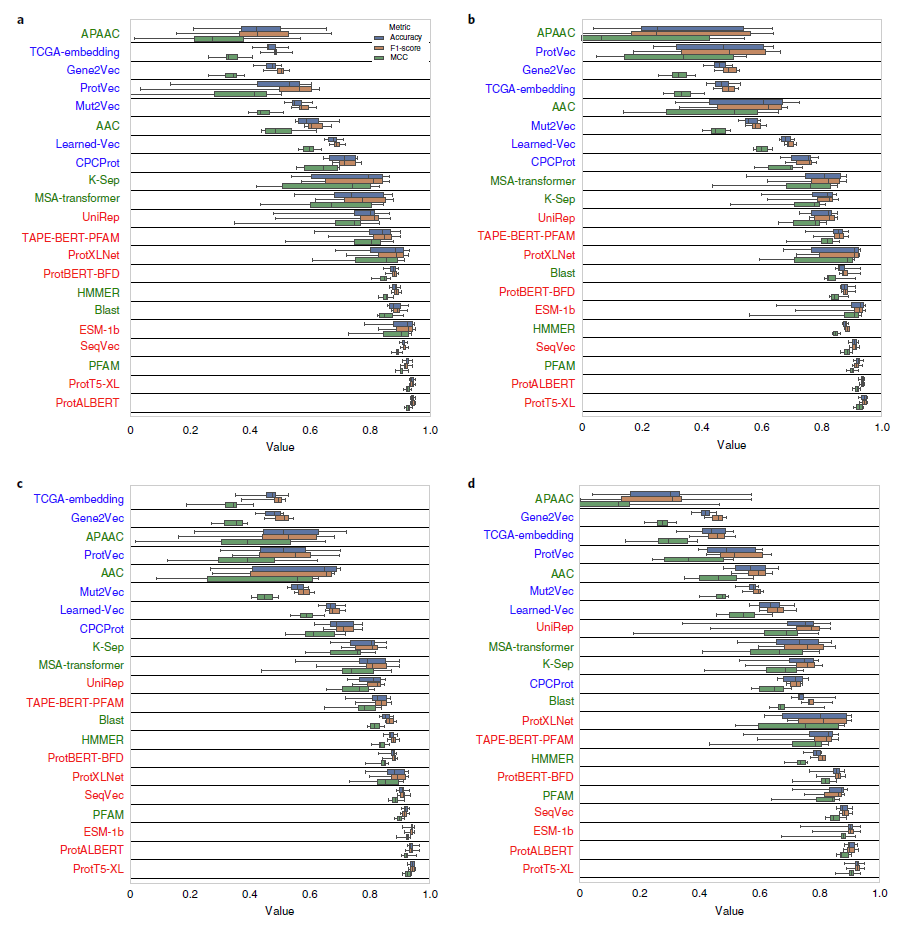
总体来看,similarity越低,结果越差。这种趋势在传统模型中比深度学习模型中表现得更明显。
twilight zone (25% similarity) 序列高于这个相似度的情况下,结构上的和功能上的annotation才能在protein间转化。换句话说,如果低于这个相似度,两个蛋白之间的结构很有可能并不相似。
尽管similarity降到15%(低于twilight zone),top representations依旧表现很好。这可能说明representation learning methods有能力捕捉简单的sequence similarity以外的信息。
蛋白质-蛋白质亲和性估计
K_D = [P][L]/[PL] [P]代表free protein的浓度(concentration) [L]代表ligand的浓度 [PL]代表complex的浓度 事实上K_D是复合物分解反应的化学平衡常数,对于可逆反应,其平衡常数是正反应的平衡常数和逆反应的平衡常数之商。即K_D=k_d/k_a。 这里之所以规定了是分解反应是因为下标,d代表disassociation,a代表association。 我们考虑的主要是蛋白细胞内的反应(intracelluar interaction),在细胞内,当反应商Q(Q=c[P]c[L]/c[PL])大于K_D,此时会倾向于逆反应,也即蛋白和受体结合;同理,当反应商Q小于K_D,此时会倾向于正反应,也即蛋白和受体分离。 因此,K_D越大,就需要越大的蛋白质浓度和受体浓度二者才能结合,说明蛋白质和受体的亲和力越差。
具体来说,是预测interaction partner部分的mutation对亲和力的改变。数据集使用的是SKEMPI,它包括了wildtype和variants的co-crystalized complexes(来自PDB)之间的PPI(Protein Protein Interaction) binding affinity score(即K_d value)